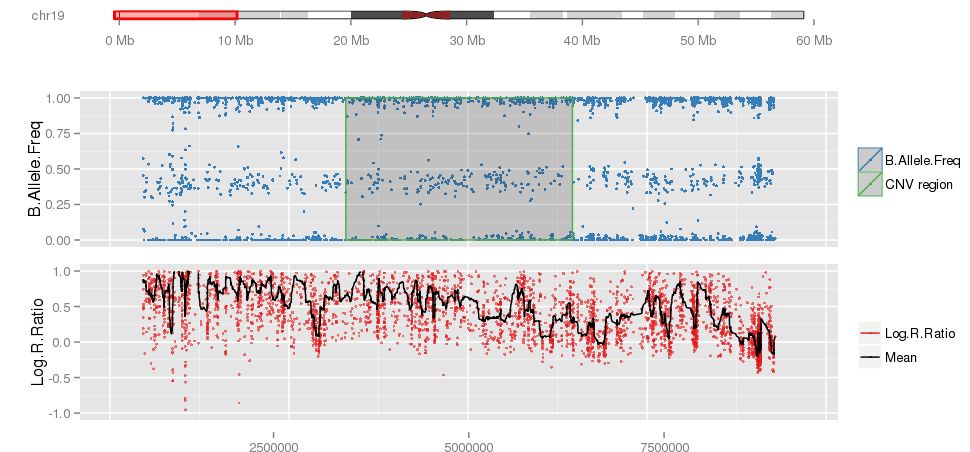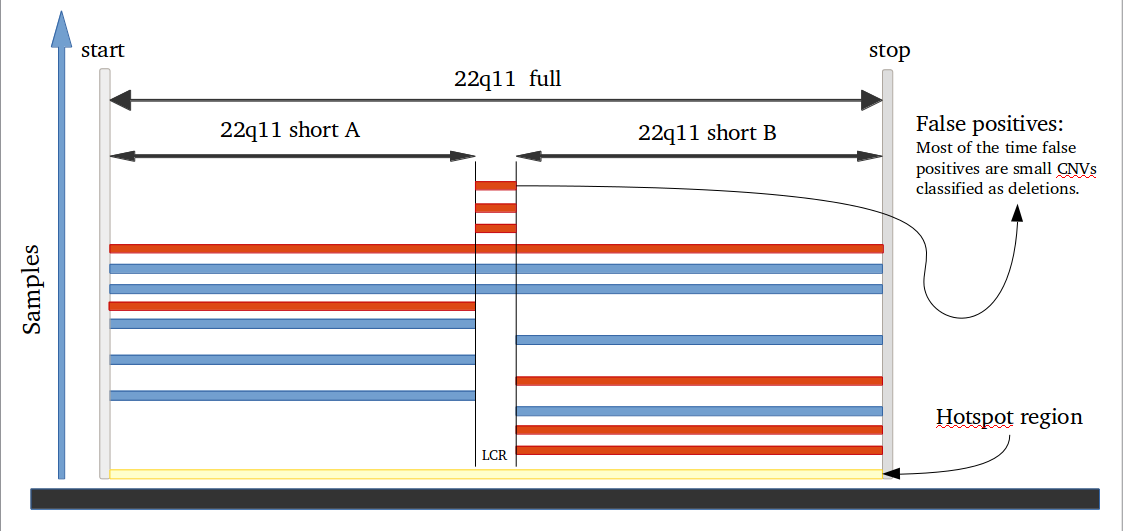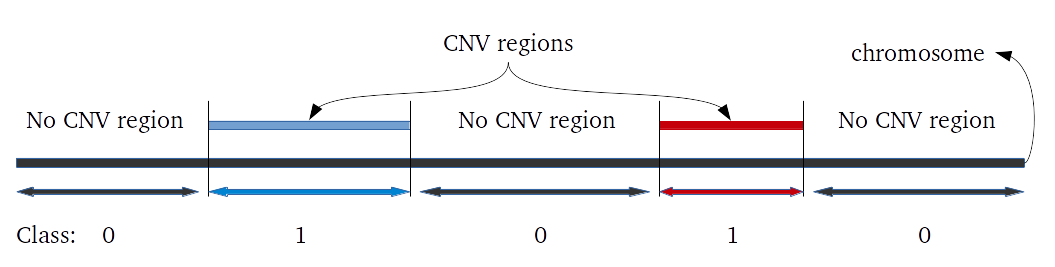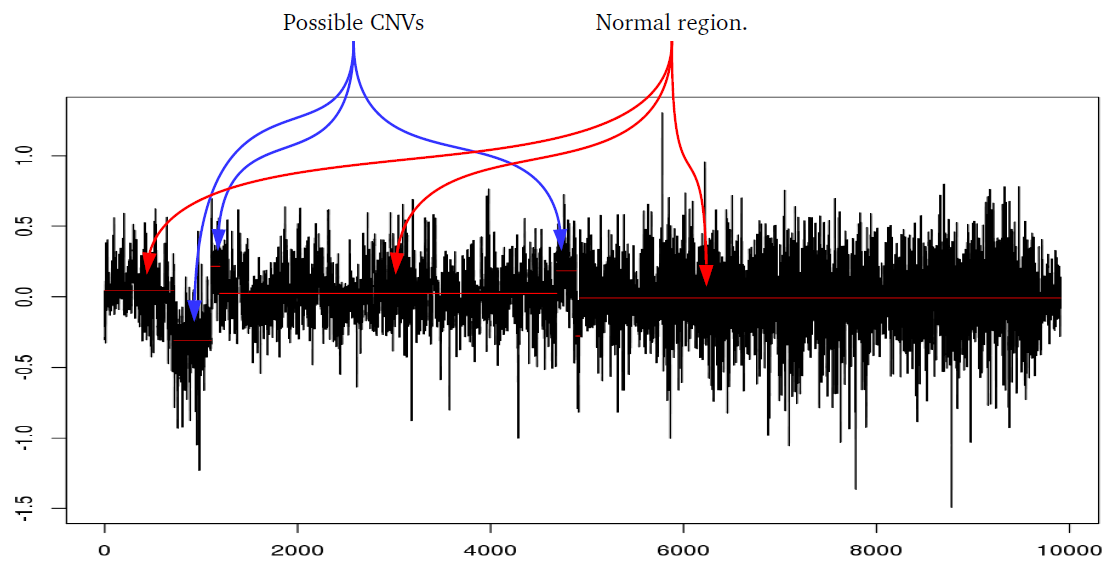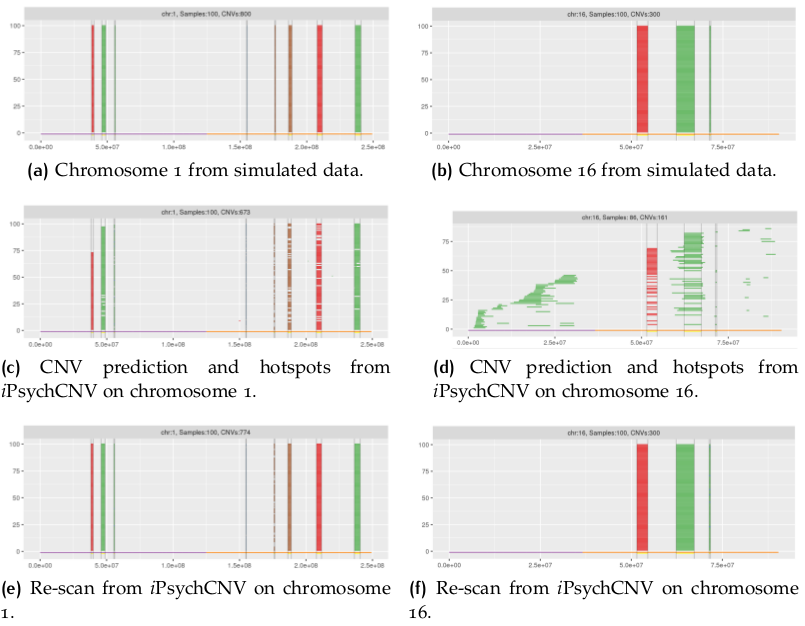
library(iPsychCNV) # Creating Mock data MockDataCNVs <- MockData(N=100, Type="Blood,", Cores=20) roi_mock <- subset(MockDataCNVs, ID %in% "MockSample_1.tab" & CN != 2) roi_mock$Class <- "ROI" tmp <- subset(MockDataCNVs, CN != 2) PlotAllCNVs(tmp, Name="MockData.png", Roi=roi_mock) # iPsychCNV prediction iPsych.Pred <- iPsychCNV(PathRawData=".", Cores=28, Pattern="^MockSample", Skip=0) PlotAllCNVs(df= iPsych.Pred, Name=" iPsych.Pred.png", Roi=roi_mock, hg="hg19") iPsych.Hotspots <- HotspotsCNV(df=iPsych.Pred, Freq=3, OverlapCutoff=0.7, Cores=22) PlotAllCNVs(df= iPsych.Pred, Name=" iPsych.Pred.png", Roi=iPsych.Hotspots, hg="hg19") iPsych.ReScan <- ReScanCNVs(CNVs=iPsych.Hotspots, PathRawData=".", Pattern="^MockSample_*", Skip=0, Cores=28, IndxPos=TRUE, CNVSignal=0, OnlyCNVs=FALSE) PlotAllCNVs(iPsych.ReScan, Name="iPsych.ReScan.png", Roi=iPsych.Hotspots)