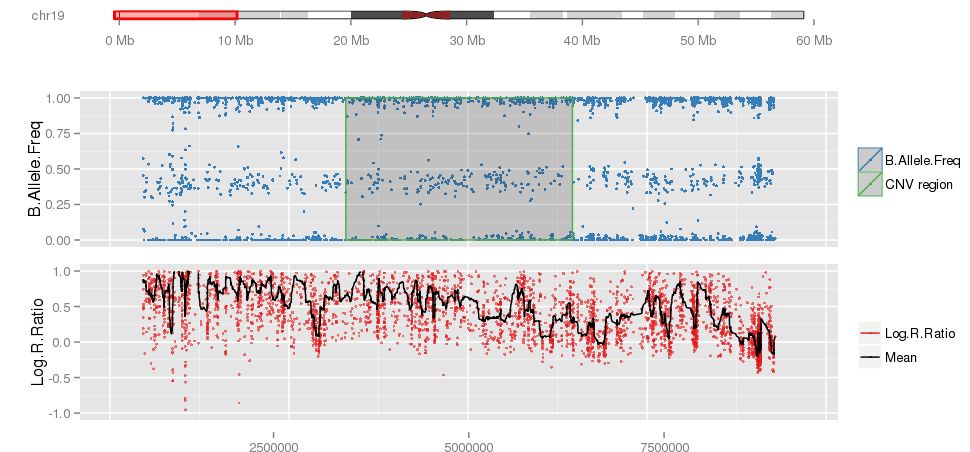
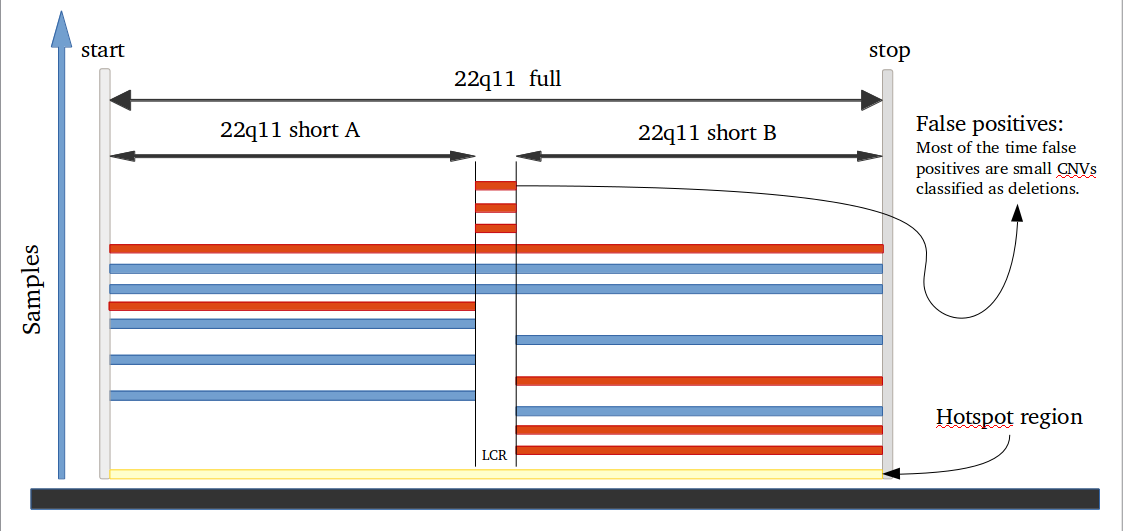
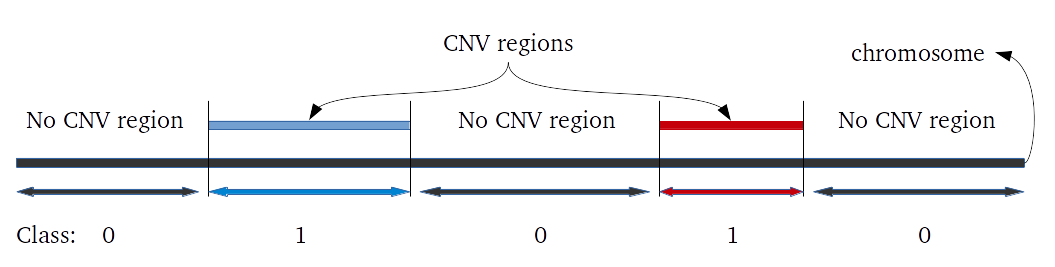
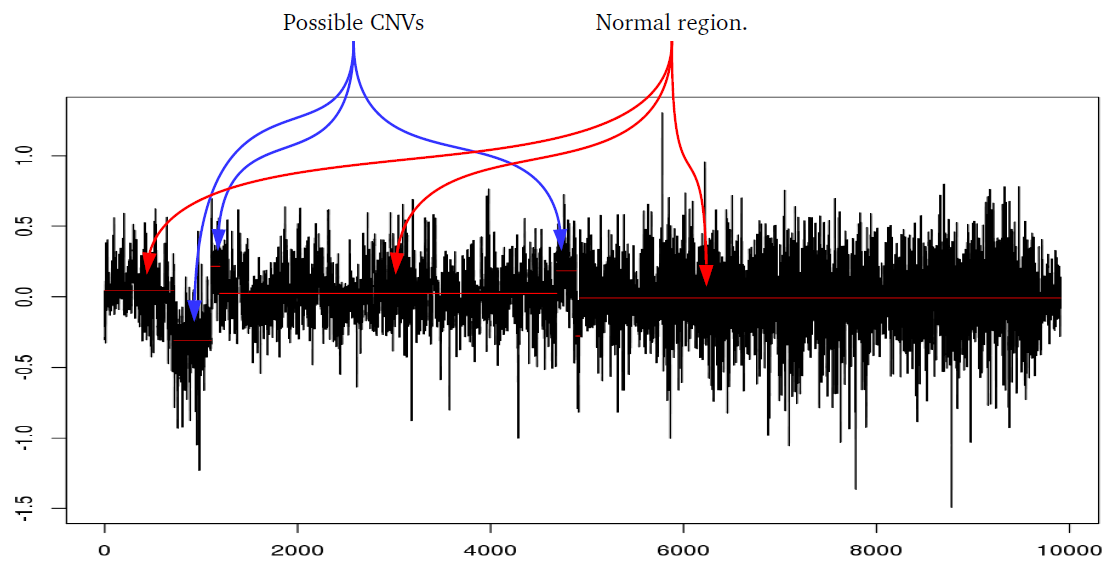
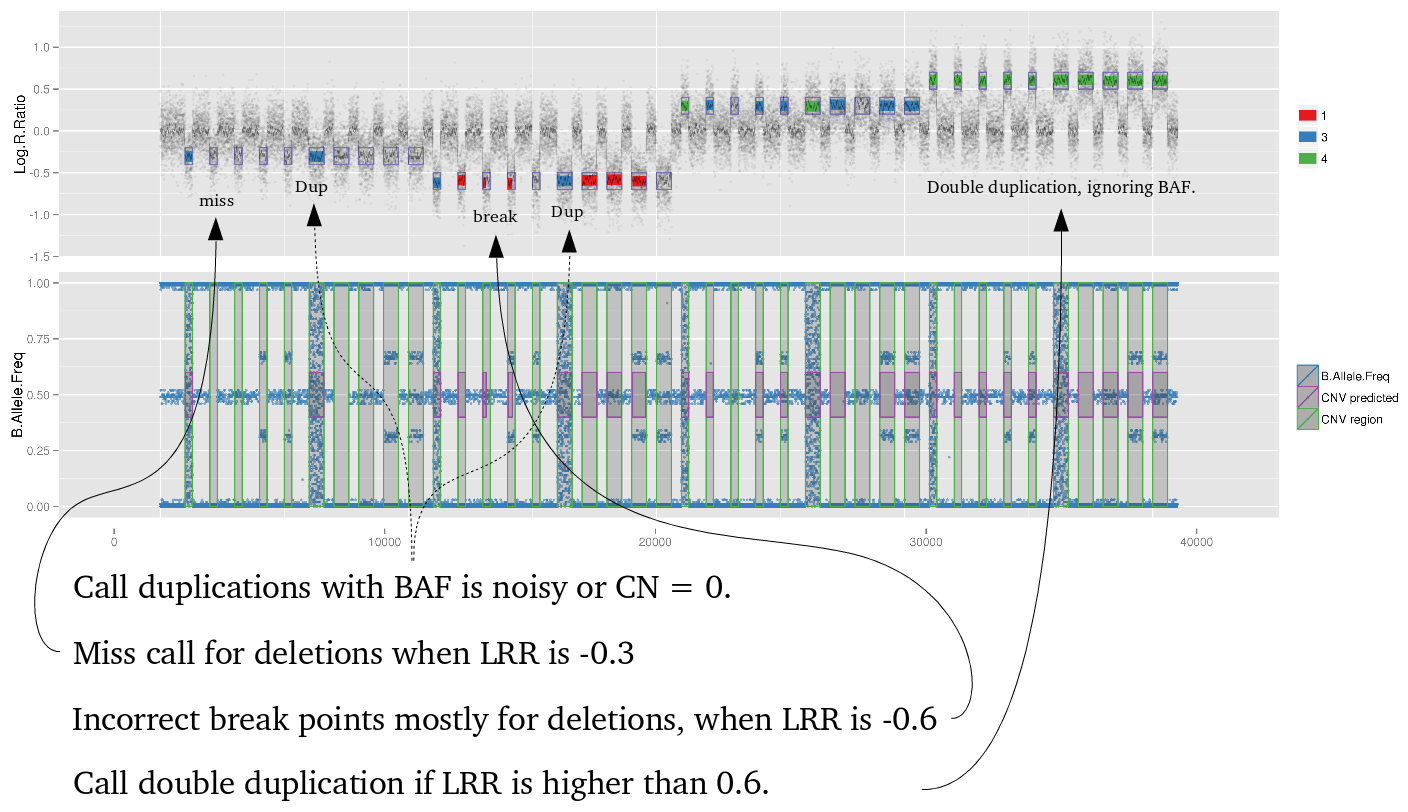
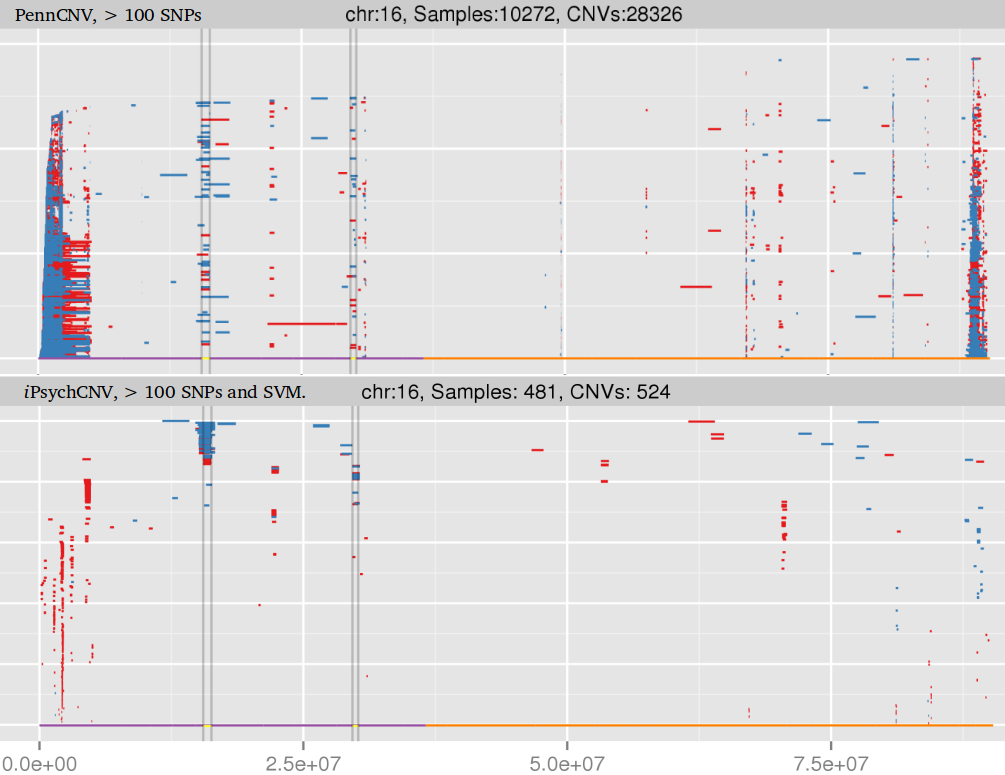
iPsychCNV R package includes a series of tools to work with CNVs from SNP arrays. Here we describe the main functions that can be used individually. The iPsychCNV function aggregates many of those tools to make the CNV call.
Read Sample
## It will read any SNP array info and return a data.frame. ## The function controls for name format and headers. ## # RawFile: The file path including the file name. # skip: If file has header, number of line to skip. # LCR: Low copy number repeats. A vector with SNP names that should be removed. # PFB: Population frequency for each SNP. If FALSE than every SNP will have PFB=0.5. # chr: A character, specifying if subset of the sample should be used. If you only want to read chr1 for example. ## Example ## MockSample.chr1 <- ReadSample(RawFile="MockSample_1.tab", skip=0, LCR=FALSE, PFB=FALSE, chr=1)Normalize Data
# Function to normalize the sample by quantile and/or QSpline. # # Sample: Sample object read from ReadSample function. # ExpectedMean: Expected mean for Log R ratio chromosome, default value zero, but it can be changed by user. # penalty: Value for smooth.spline function from stats package, default value 60. Only valid if QSpline is TRUE. # QSpline: TRUE or FALSE for smooth.spline function. # Quantile: TRUE or FALSE for quantile normalization. Uses normalize.quantiles function from preprocessCore package. # sd: Standard deviation for fake samples chromosome, with mean equal 0, default value 0.18. ## Example ## Sample.norm <- NormalizeData(Sample=MockSample.chr1, ExpectedMean=0, penalty=60, QSpline=FALSE, Quantile=FALSE, sd=0.18)Find CNVs
# Function to find CNVs start and stop position using PELT method. # # ID: Sample name # Sample: Sample object read from ReadSample function. # CPTmethod: Define which method to find change points from changepoint package. Default value "meanvar" or "mean". # CNVSignal: Minimum Log R Ratio signal from change point to be considered a CNV, default value 0.1 (CNVs with mean LRR # -0.1 > and < 0.1 are not considered). # penvalue: pen.value option for cpt.meanvar, only used if CPTmethod = "meanvar". ## Example ## CNVs <- FindCNV.V4(ID="Sample.Name", Sample=Sample, CPTmethod="meanvar", CNVSignal=0.1, penvalue=10)Merge CNVs
# Function to merge CNVs. It is integrated in Find CNVs function, but can be used separately. # df: Data frame result from Find CNV function. # MaxNumSNPs: Determinate if two CNVs should be merged. If two CNVs are apart for more then MaxNumSNPs it will not be # merged. Default value 50 SNPs. ## Example ## CNVs.merged <- MergeCNVs(df=CNVs, MaxNumSNPs=50)Filter CNVs
# Function to filter CNVS. # CNVs: Data frame result from Find CNV function. # MinNumSNPs: Minimum number of SNPs in a CNV. # Sample: Sample object read from ReadSample function. # ID: Sample name. ## Example ## CNVs.Good <- FilterCNVs.V4(CNVs=CNVs, MinNumSNPs=20, Sample=Sample, ID="SampleName")Hotspots
# A function that will combine CNVs that overlap with given specific cutoffs. # df: Data frame result from filter CNV function. # Freq: Minimum number of CNVs in the hotspots to be accepted, default value 1. # OverlapCutoff: Percentage of CNV length overlap on both ways to be accepted in a hotspot, default value 0.7 (70%). # Cores: Number of CPUs to be used if multiple cpus are available, default value 1. ## Example ## CNVs.Hotspots <- HotspotsCNV(df=CNVs.Good, Freq=1, OverlapCutoff=0.7, Cores=22)Evaluate CNVs
# Evaluating CNVs prediction performance. # MockCNVs: a data frame with the known CNVs from a mock data. It can be from long mock or mock data. # df: data frame with CNVs predicted. ## Example ## CNVs.Eval <- EvaluateMockResults(LongRoi, PredictedCNV)Re-Scan CNVs
# Function that will re-scan the samples searching for CNVs in specific regions (e.g. Hotspots) # CNVs: Data frame result from HotspotsCNV function. # PathRawData: Path for all samples. # MINNumSNPs: Minimum number of SNPs in a CNV, default value 20. # Cores: Number of CPUs to be used if multiple cpus are available, default value 1. # hg: Human genome build, default value "hg18" or "hg19" # NumFiles: Total number of files to be analysed. # Pattern: Pattern for sample name. Example: "*.txt" # MinLength: Minimum base pair length for CNV, default value 10. # SelectedFiles: Vector with samples's name. It will only process those files. # Skip: integer: the number of lines of the data file to skip before beginning to read data. # LCR: Low copy number repeats. A vector with SNP names that should be removed. # PFB: A file with the population frequency for each SNP. Default value NULL (every SNP with 0.5). # chr: Character if only a specific chromosome should be analysed. Example chr="1", or chr=c("1","2"), # default value NA (all chromosomes). # penalty: Value for smooth.spline function from stats package, default value 60. Only valid if QSpline is TRUE. # QSpline: TRUE or FALSE for smooth.spline function. # Quantile: TRUE or FALSE for quantile normalization. Uses normalize.quantiles function from preprocessCore package. # sd: Standard deviation for fake samples chromosome, with mean equal 0, default value 0.18. # recursive: Logical. from list.files function. Should the listing recurse into directories ? # CPTmethod: Define which method to find change points from changepoint package. Default value "meanvar" or "mean". # CNVSignal: Minimum Log R Ratio signal from change point to be considered a CNV, default value 0.1 (CNVs with mean LRR # -0.1 > and < 0.1 are not considered). # penvalue: pen.value option for cpt.meanvar, only used if CPTmethod = "meanvar". # OutputPath: Path for output results, if results should be written in a file. Default value NA, it will only return # results as an R object. ## Example ## CNVs.rescan <- ReScanCNVs(CNVs=CNVs, PathRawData = "/media/NeoScreen/NeSc_home/ILMN/iPSYCH/", MINNumSNPs=20, Cores=1, hg="hg18", NumFiles="All", Pattern="22q11_*", MinLength=10, SelectedFiles=NA, Skip=10, LCR=FALSE, PFB=NULL, chr=NA, penalty=60, Quantile=FALSE, QSpline=FALSE, sd=0.18, recursive=FALSE, CPTmethod="meanvar", CNVSignal=0.1, penvalue=10, OutputPath=NA)Check for Abnormal Chromosomes
# Function to check abnormal chromosomes. Most of methods to detect CNVs fail to detect large chromosomal events. # Path2RawFiles: Path for samples. # Cores: Number of CPUs to be used if multiple cpus are available, default value 1. # Pattern: Pattern for sample name. Example: "*.txt" # Skip: integer: the number of lines of the data file to skip before beginning to read data. # NumFiles: Total number of files to be analysed. ## Example ## AbnChrs <- CheckAbnChr(Path2RawFiles="/media/NeoScreen/NeSc_home/ILMN/iPSYCH/", Cores=40, Pattern="*.txt$", skip=10, NumFiles="All")